


Curr. Oncol. 2024, 31(4), 2341-2352; https://doi.org/10.3390/curroncol31040174 - 21 Apr 2024
Abstract
This review of the palliation of various gastro-intestinal (GI) symptoms encountered in cancer patients is by no means exhaustive. Frequent symptoms such as constipation, nausea and vomiting, bowel obstructions, ascites and bleeds will be discussed, focusing on their assessment and most importantly, how
[...] Read more.
This review of the palliation of various gastro-intestinal (GI) symptoms encountered in cancer patients is by no means exhaustive. Frequent symptoms such as constipation, nausea and vomiting, bowel obstructions, ascites and bleeds will be discussed, focusing on their assessment and most importantly, how to control the associated symptoms. All of these symptoms and GI complications can significantly impact patients’ quality of life (QOL) and should be treated as quickly and aggressively as possible.
Full article
(This article belongs to the Special Issue 2023–2024 Article Series of the Canadian Association of General Practitioners in Oncology)

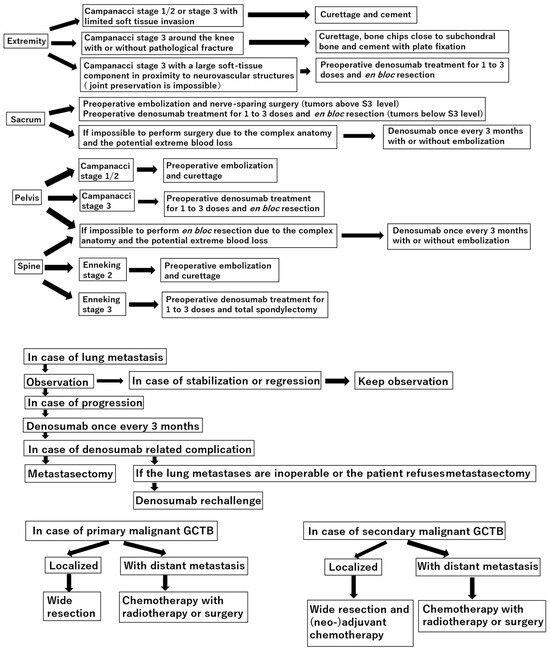





{kind=link}
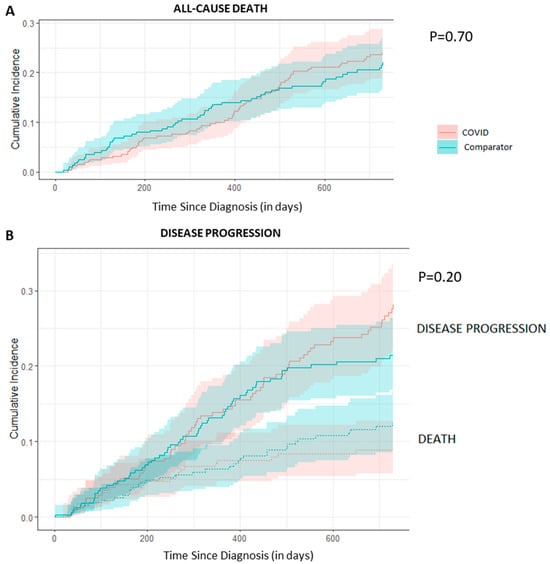
{kind=link}
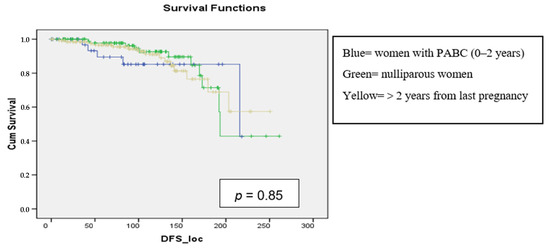
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
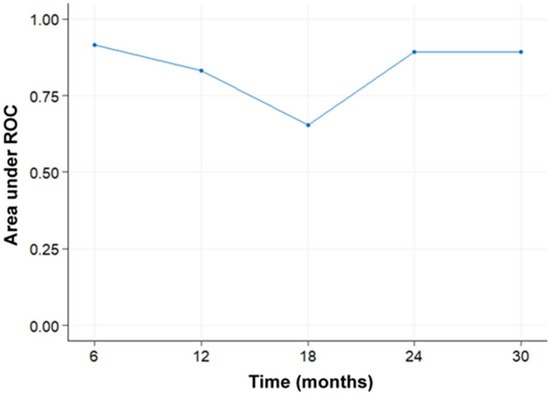{kind=link}
{kind=link}
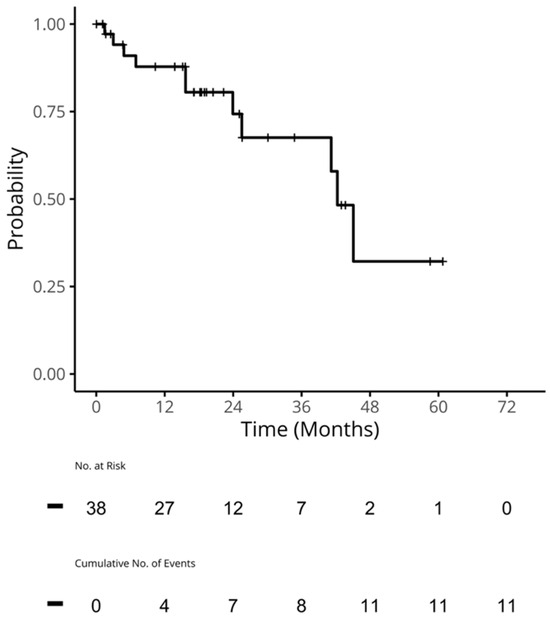{kind=link}
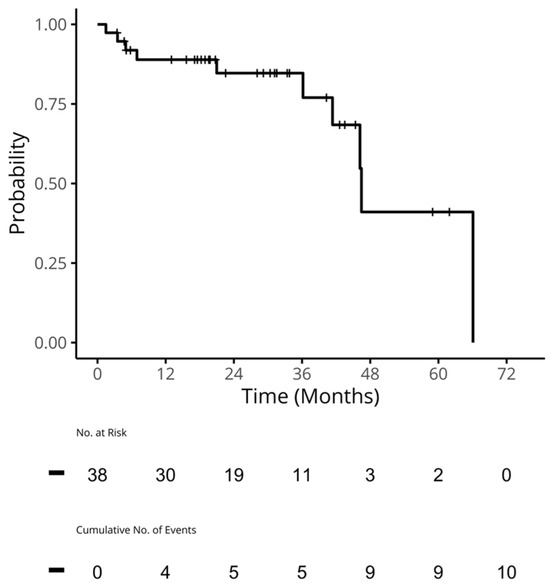{kind=link}
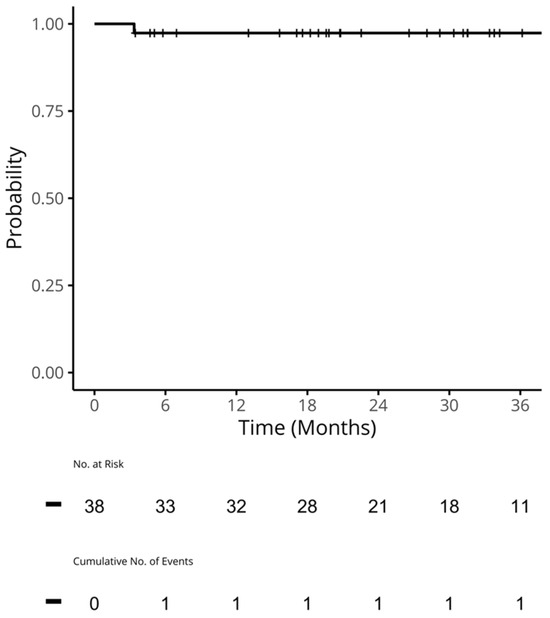{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}