


Curr. Oncol. 2024, 31(4), 2328-2340; https://doi.org/10.3390/curroncol31040173 - 19 Apr 2024
Abstract
We undertook a retrospective study to compare the quality of care delivered to a cohort of newly diagnosed adults with colon, rectal or anal cancer during the early phase of COVID-19 (02/20–12/20) relative to the same period in the year prior (the comparator
[...] Read more.
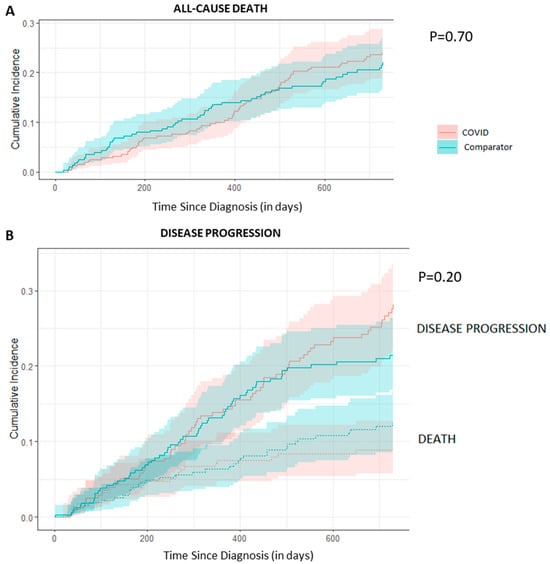
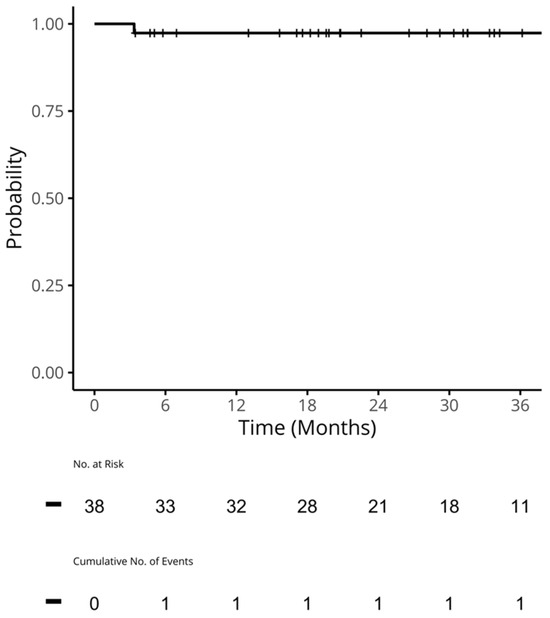
We undertook a retrospective study to compare the quality of care delivered to a cohort of newly diagnosed adults with colon, rectal or anal cancer during the early phase of COVID-19 (02/20–12/20) relative to the same period in the year prior (the comparator cohort), and examine the impact of the pandemic on 2-year disease progression and all-cause mortality. We observed poorer performance on a number of quality measures, such as approximately three times as many patients in the COVID-19 cohort experienced 30-day post-surgical readmission (10.5% vs. 3.6%; SD:0.27). Despite these differences, we observed no statistically significant adjusted associations between COVID-19 and time to either all-cause mortality (HR: 0.88, 95% CI: 0.61–1.27, p = 0.50) or disease progression (HR: 1.16, 95% CI: 0.82–1.64, p = 0.41). However, there was a substantial reduction in new patient consults during the early phase of COVID-19 (12.2% decrease), which appeared to disproportionally impact patients who traditionally experience sociodemographic disparities in access to care, given that the COVID-19 cohort skewed younger and there were fewer patients from neighborhoods with the highest Housing and Dwelling, ands Age and Labour Force marginalization quintiles. Future work is needed to understand the more downstream effects of COVID-19 related changes on cancer care to inform planning for future disruptions in care.
Full article
(This article belongs to the Section Gastrointestinal Oncology)
►
Show Figures
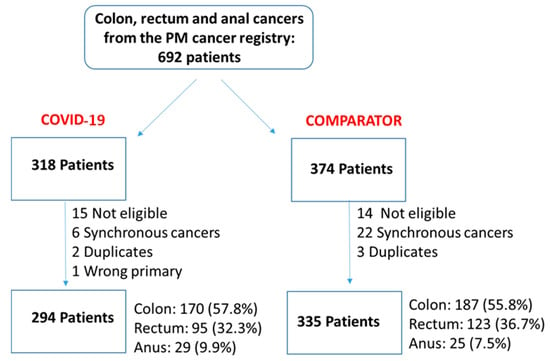
Figure 1






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}