


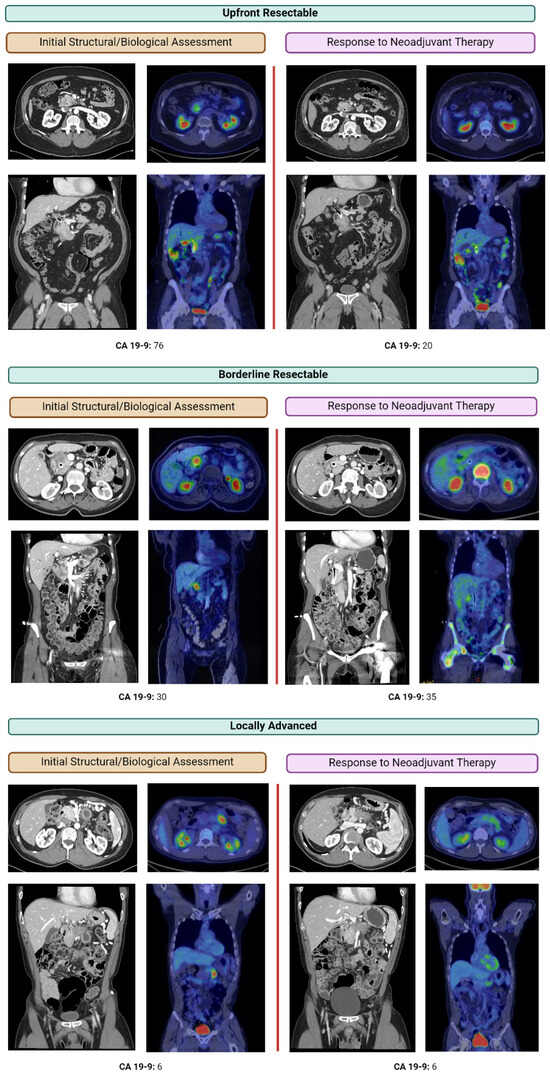
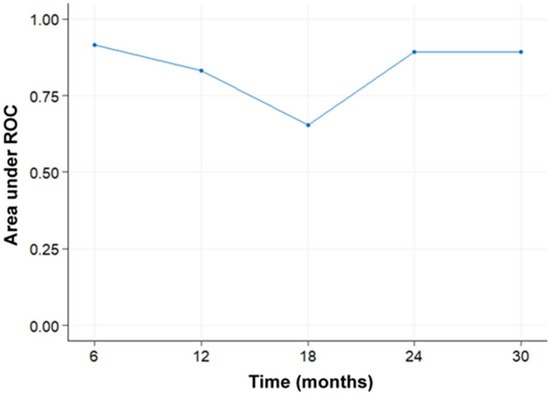
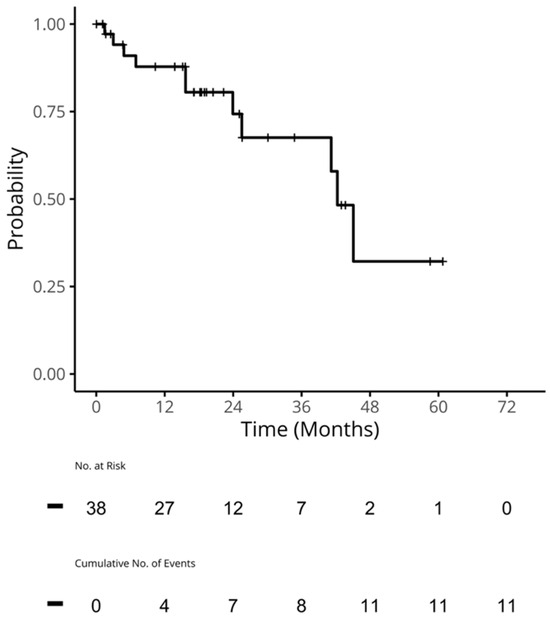
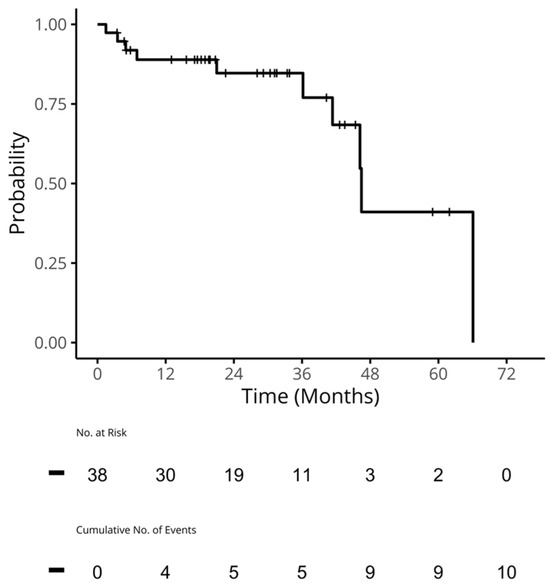
Curr. Oncol. 2024, 31(5), 2410-2419; https://doi.org/10.3390/curroncol31050180 (registering DOI) - 24 Apr 2024
Abstract
Therapeutic management of patients with leptomeningeal carcinomatosis (LC) may require treatment of concomitant hydrocephalus (HC) in addition to intrathecal chemotherapy (ITC). Ventriculoperitoneal shunts (VPS) equipped with a valve for manual deactivation of shunt function and a concomitant reservoir for application of ITC pose
[...] Read more.
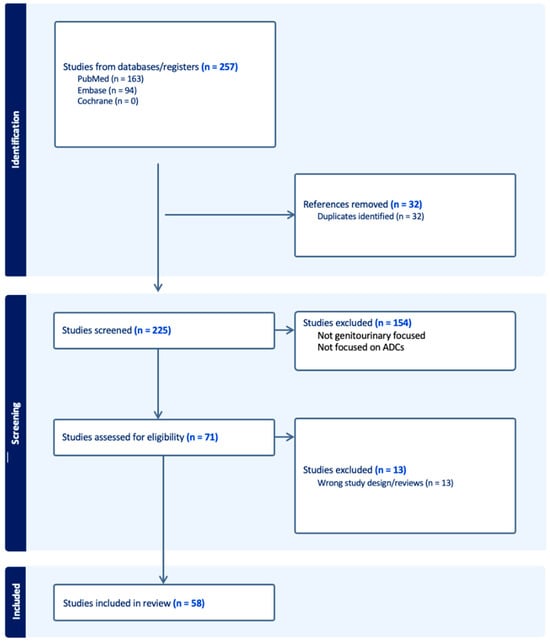
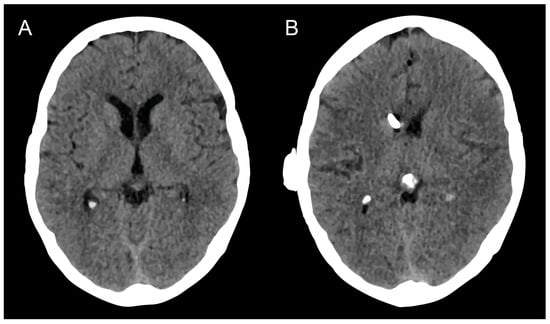
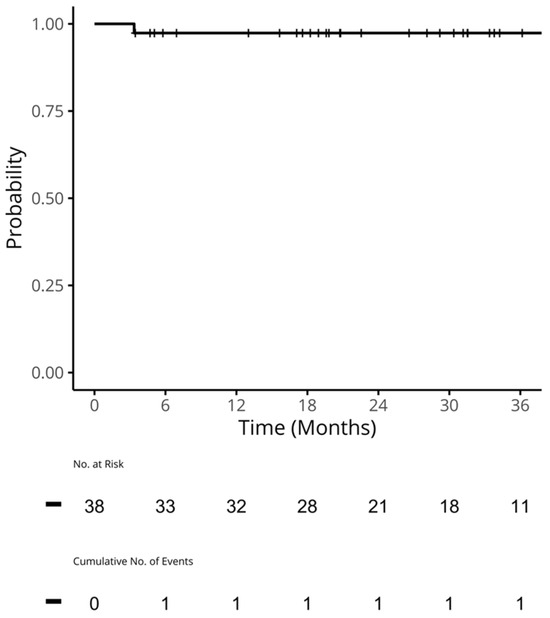
Therapeutic management of patients with leptomeningeal carcinomatosis (LC) may require treatment of concomitant hydrocephalus (HC) in addition to intrathecal chemotherapy (ITC). Ventriculoperitoneal shunts (VPS) equipped with a valve for manual deactivation of shunt function and a concomitant reservoir for application of ITC pose an elegant solution to both problems. The present study evaluates indication, feasibility, and safety of such a modified shunt/reservoir design (mS/R). All patients with LC aged ≥ 18 years who had undergone mS/R implantation between 2013 and 2020 at the authors’ institution were further analyzed. ITC was indicated following the recommendation of the neuro-oncological tumor board and performed according to a standardized protocol. Sixteen patients with LC underwent mS/R implantation for subsequent ITC and concomitant treatment of HC. Regarding HC-related clinical symptoms, 69% of patients preoperatively exhibited lethargy, 38% cognitive impairment, and 38% (additional) visual disturbances. Postoperatively, 86% of patients achieved subjective improvement of HC-related symptoms. Overall, postoperative complications occurred in three patients (19%). No patient encountered cancer treatment-related complications. The present study describes a combination procedure consisting of a standard VPS-system and a standard reservoir for patients suffering from LC and HC. No cancer treatment-related complications occurred, indicating straightforward handling and thus safety.
Full article
(This article belongs to the Section Neuro-Oncology)
►
Show Figures
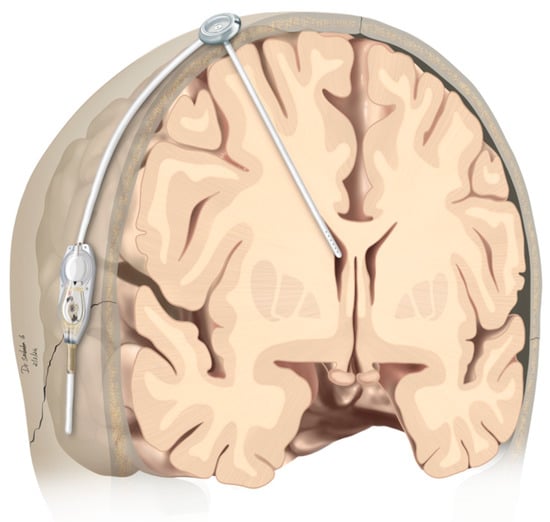
Figure 1





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}