


Curr. Oncol. 2024, 31(5), 2364-2375; https://doi.org/10.3390/curroncol31050176 (registering DOI) - 23 Apr 2024
Abstract
Background: Breast cancer (BC) is frequently diagnosed among Canadian women. While targeted therapies are available for most BC patients; treatment resistance is common and novel therapeutic targets are of interest. Thyroid hormones (TH) bound to thyroid hormone receptors (THR) influence cell proliferation and
[...] Read more.
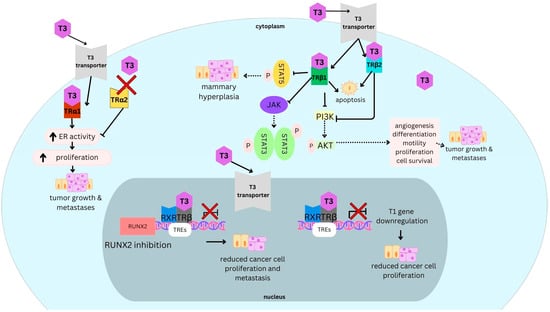
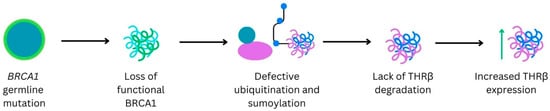
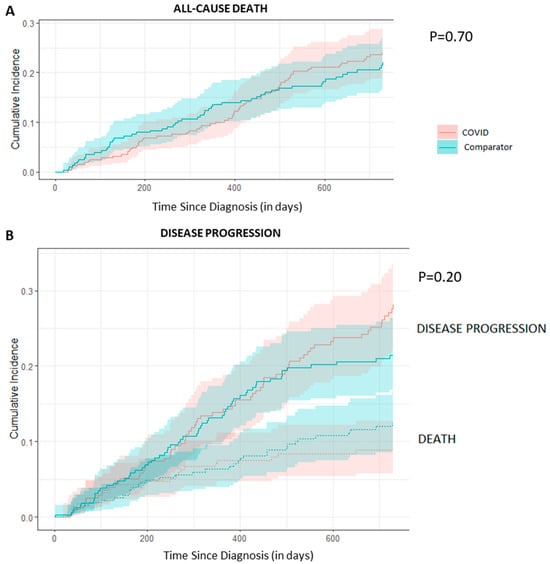
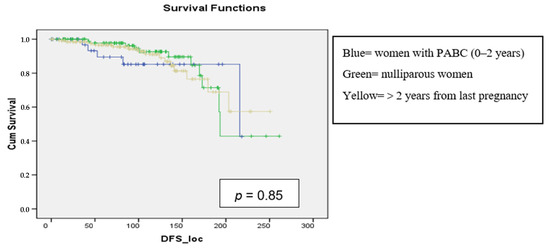
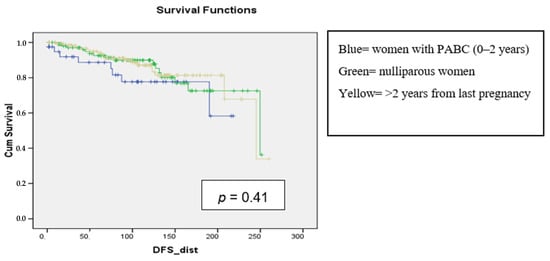
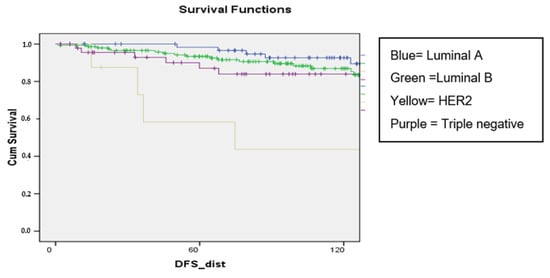
Background: Breast cancer (BC) is frequently diagnosed among Canadian women. While targeted therapies are available for most BC patients; treatment resistance is common and novel therapeutic targets are of interest. Thyroid hormones (TH) bound to thyroid hormone receptors (THR) influence cell proliferation and differentiation; they are also involved in the growth and development of normal breast tissue. Evidence suggests that THRβ is a tumor suppressor in various solid tumors. Purpose: This narrative review discusses retrospective studies regarding the clinical relevance of THRβ as a potential prognostic biomarker and therapeutic target in BC. Methods: We consulted with an information specialist to develop a search strategy to find all literature related to THRα expression as a potential prognostic and therapeutic biomarker in breast cancer. The primary search was developed for Medline and translated to Embase. The searches were conducted on the Ovid platform on 18 August 2023. Results: Across seven retrospective studies identified, several have shown an association between higher THRβ1 expression with a lower risk of BC recurrence and with longer overall survival. Conclusions: Some evidence suggests that THRβ expression is associated with a lower risk of BC recurrence and death. Validation of THRβ as an independent prognostic biomarker and possible predictive biomarker of response to endocrine therapy and/or chemotherapy is of interest. Given that THRβ is upstream of the AKT/PI3K pathway, its potential as a predictive biomarker of response to AKT inhibitors and/or PI3K inhibitors may also be of value. Finally, the potential re-purposing of THRβ agonists as anti-cancer agents warrants investigation.
Full article
(This article belongs to the Special Issue Advanced Approaches to Breast Cancer Biomarkers)
►
Show Figures
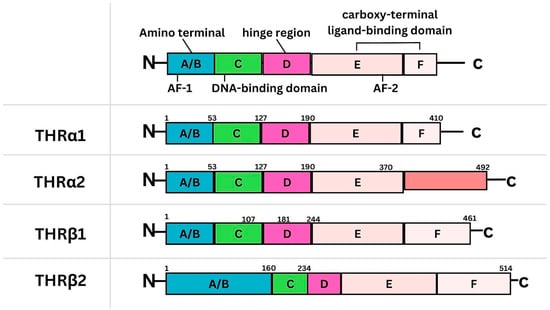
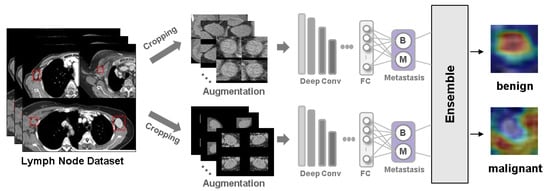
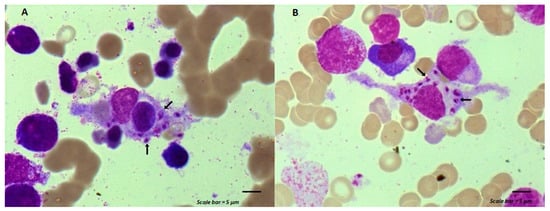
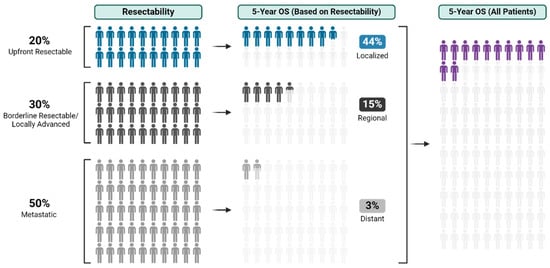
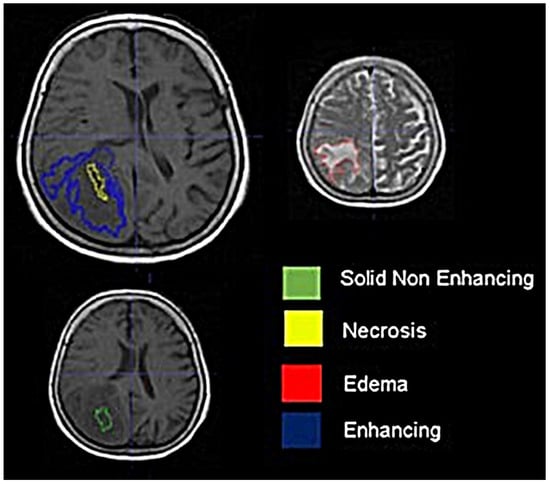
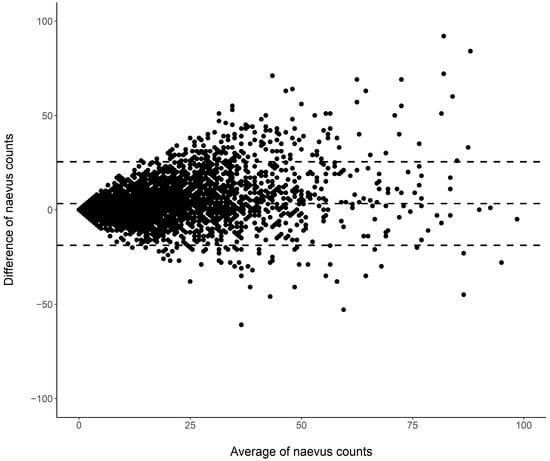
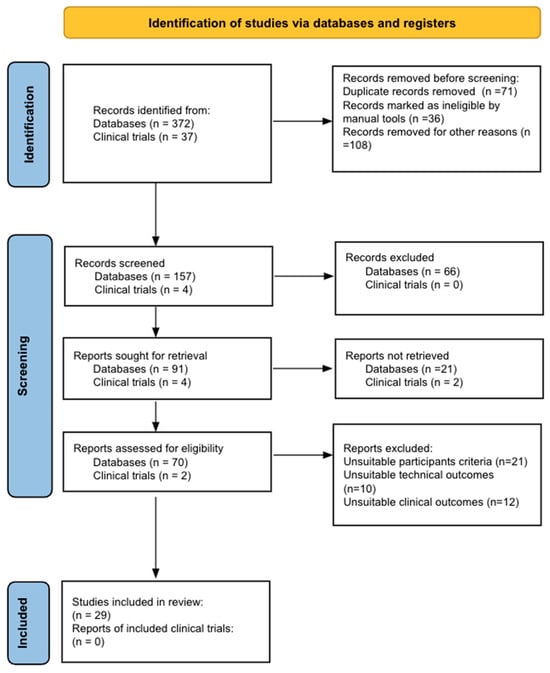
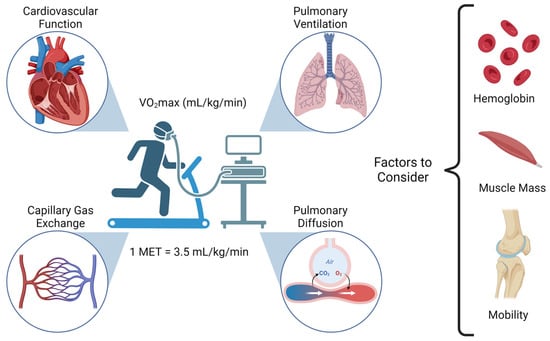
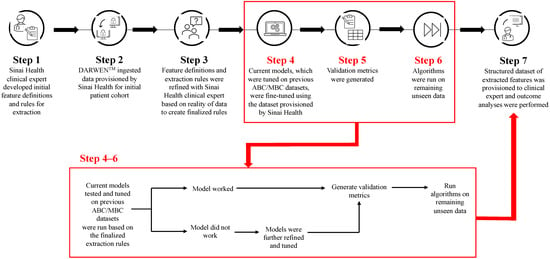
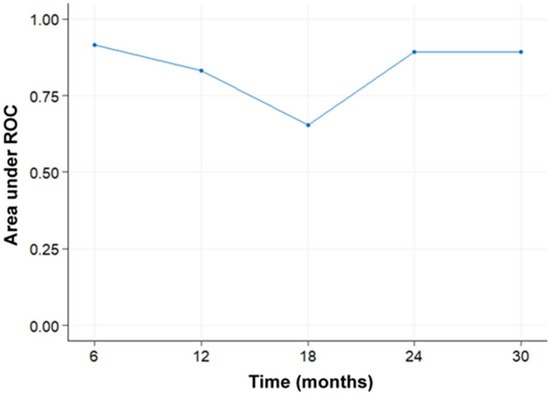
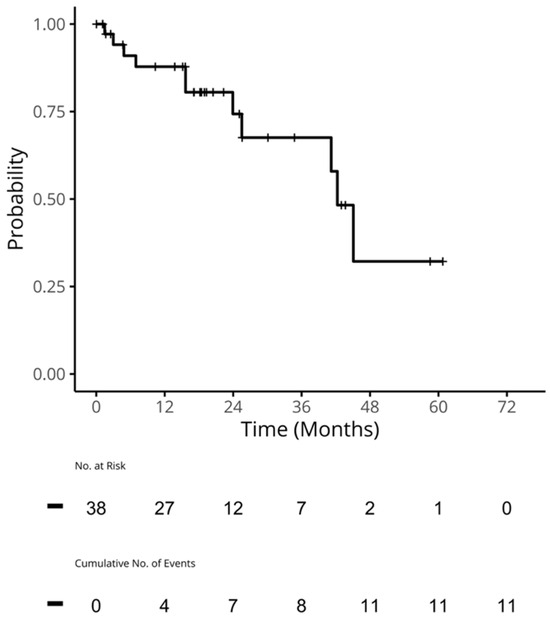
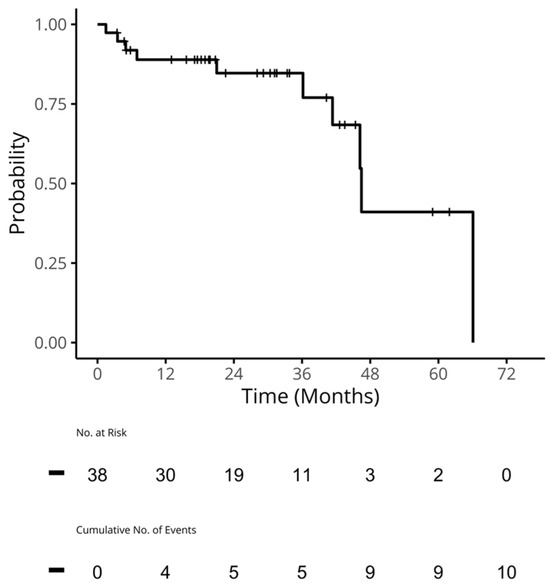
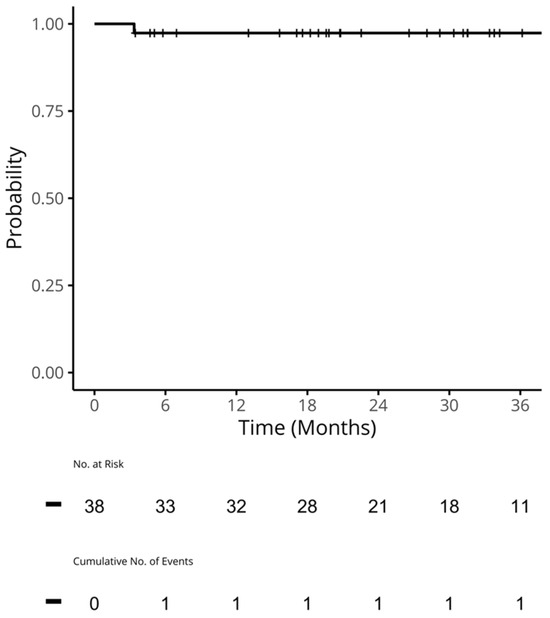
Figure 1





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}