


Curr. Oncol. 2024, 31(4), 2316-2327; https://doi.org/10.3390/curroncol31040172 - 19 Apr 2024
Abstract
The treatment landscape of genitourinary cancers has significantly evolved over the past few years. Renal cell carcinoma, bladder cancer, and prostate cancer are the most common genitourinary malignancies. Recent advancements have produced new targeted therapies, particularly antibody–drug conjugates (ADCs), due to a better
[...] Read more.
The treatment landscape of genitourinary cancers has significantly evolved over the past few years. Renal cell carcinoma, bladder cancer, and prostate cancer are the most common genitourinary malignancies. Recent advancements have produced new targeted therapies, particularly antibody–drug conjugates (ADCs), due to a better understanding of the underlying oncogenic factors and molecular mechanisms involved. ADCs function as a ‘drug delivery into the tumor’ system. They are composed of an antigen-directed antibody linked to a cytotoxic drug that releases cytotoxic components after binding to the tumor cell’s surface antigen. ADCs have been proven to be extremely promising in the treatment of several cancer types. For GU cancers, this novel treatment has only benefited patients with metastatic urothelial cancer (mUC). The rest of the GU cancer paradigm does not have any FDA-approved ADC treatment options available yet. In this study, we have thoroughly completed a narrative review of the current literature and summarized preclinical studies and clinical trials that evaluated the utility, activity, and toxicity of ADCs in GU cancers, the prospects of ADC development, and the ongoing clinical trials. Prospective clinical trials, retrospective studies, case reports, and scoping reviews were included.
Full article
(This article belongs to the Special Issue The Evolving Role of Antibody Drug Conjugates in Cancer Therapy)
►
Show Figures
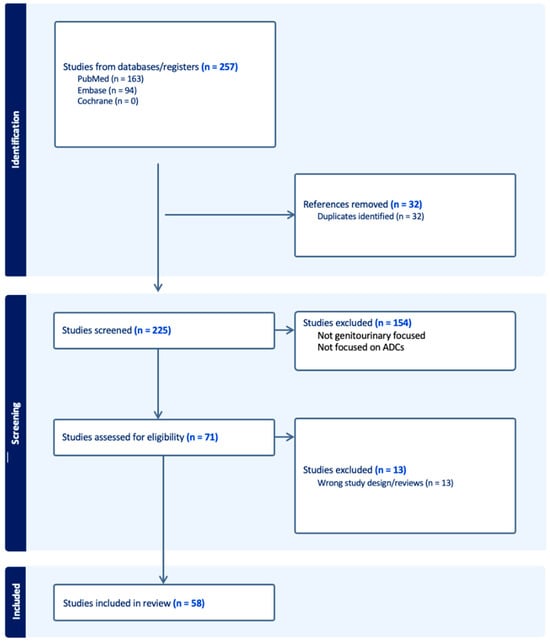
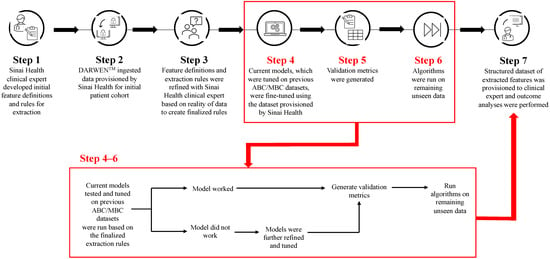
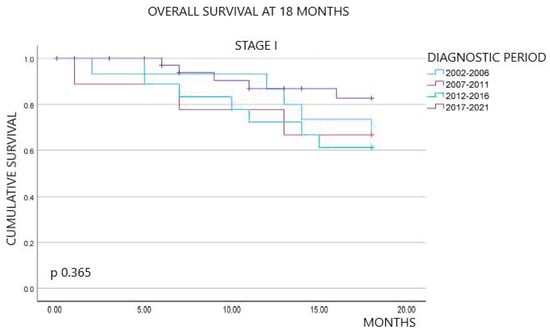
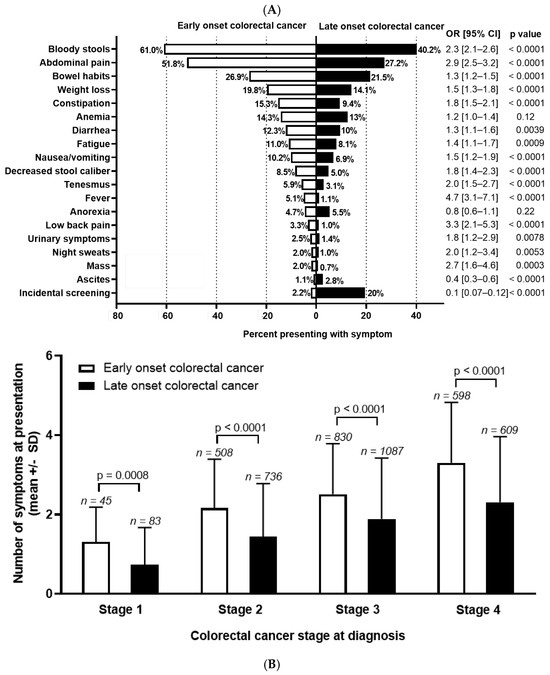
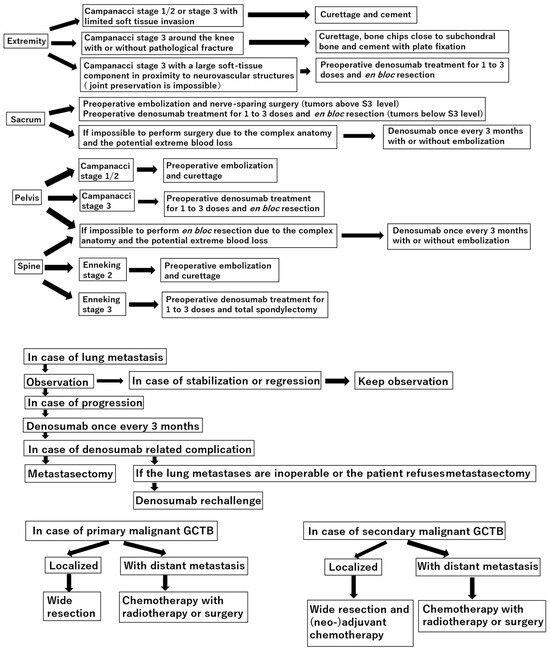
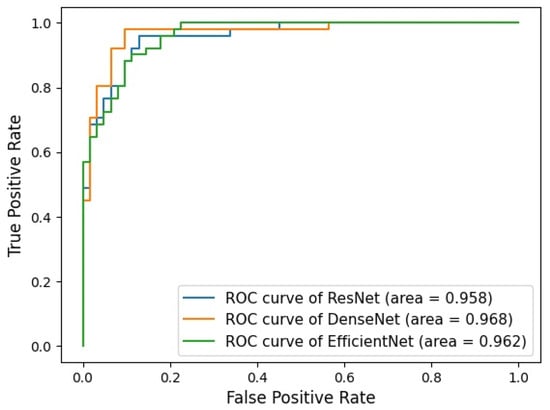
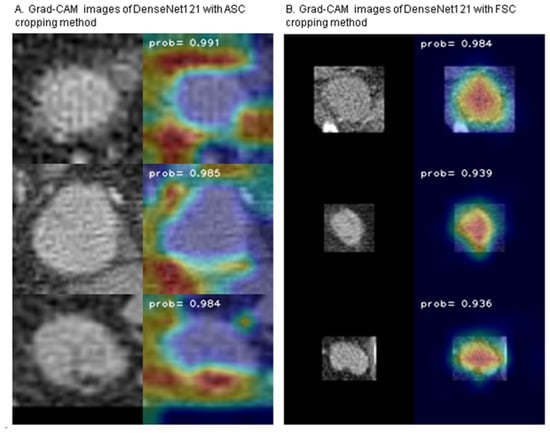
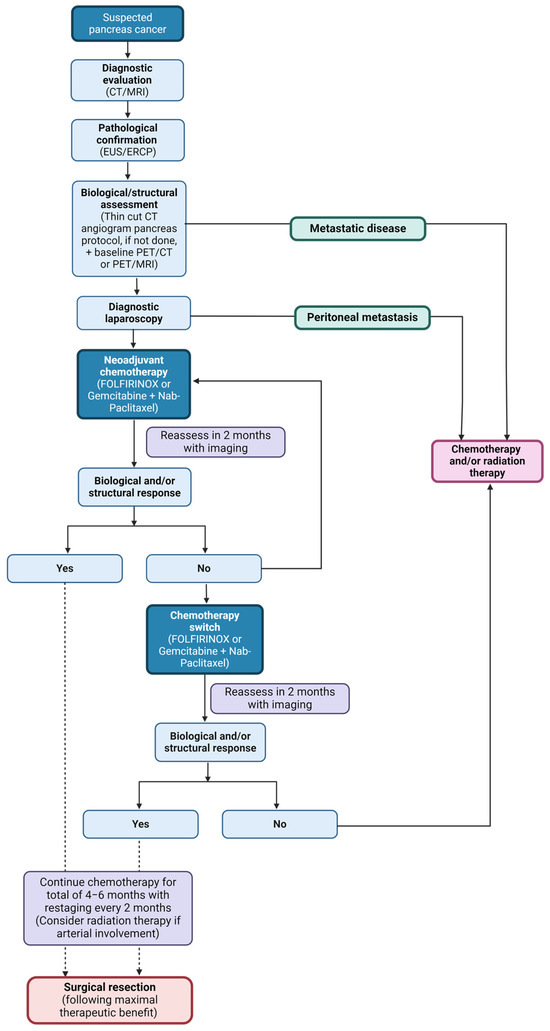
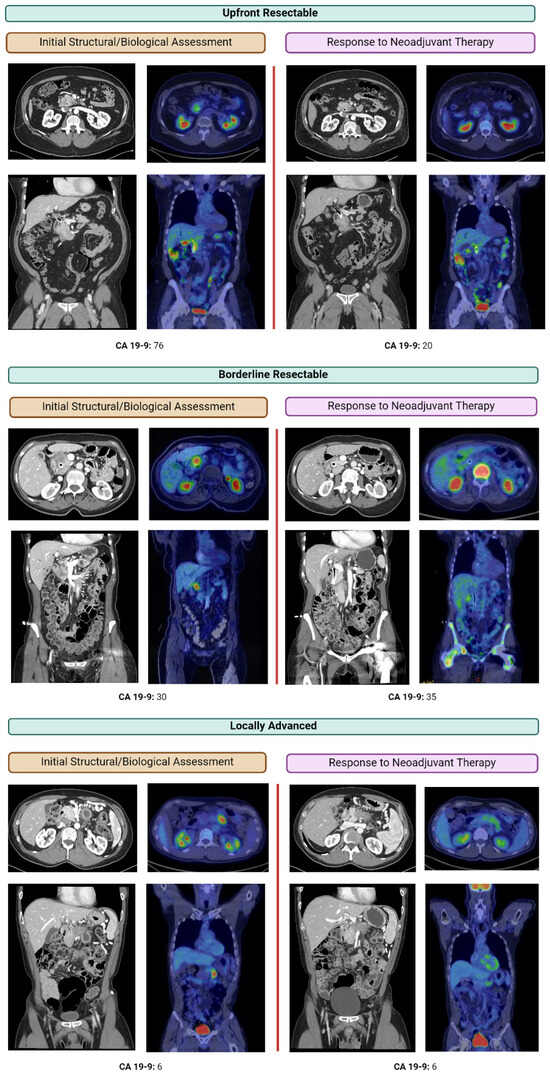
Figure 1






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}