


Curr. Oncol. 2024, 31(4), 2260-2273; https://doi.org/10.3390/curroncol31040167 - 15 Apr 2024
Abstract
With an overall 5-year survival rate of 12%, pancreas ductal adenocarcinoma (PDAC) is an aggressive cancer that claims more than 50,000 patient lives each year in the United States alone. Even those few patients who undergo curative-intent resection with favorable pathology reports are
[...] Read more.
With an overall 5-year survival rate of 12%, pancreas ductal adenocarcinoma (PDAC) is an aggressive cancer that claims more than 50,000 patient lives each year in the United States alone. Even those few patients who undergo curative-intent resection with favorable pathology reports are likely to experience recurrence within the first two years after surgery and ultimately die from their cancer. We hypothesize that risk factors for these early recurrences can be identified with thorough preoperative staging, thus enabling proper patient selection for surgical resection and avoiding unnecessary harm. Herein, we review evidence supporting multidisciplinary and multimodality staging, comprehensive neoadjuvant treatment strategies, and optimal patient selection for curative-intent surgical resections. We further review data generated from our standardized approach at the Mayo Clinic and extrapolate to inform potential future investigations.
Full article
(This article belongs to the Special Issue New Treatments in Pancreatic Ductal Adenocarcinoma)
►
Show Figures
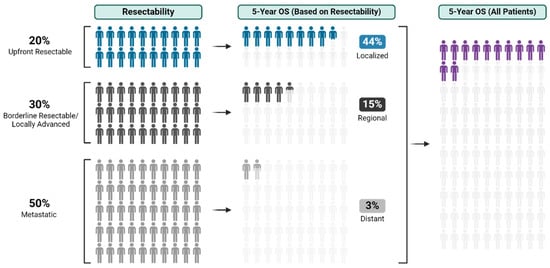
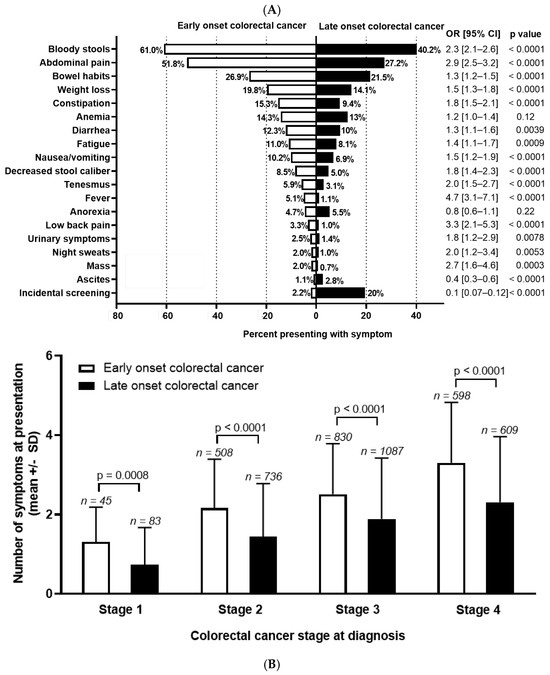
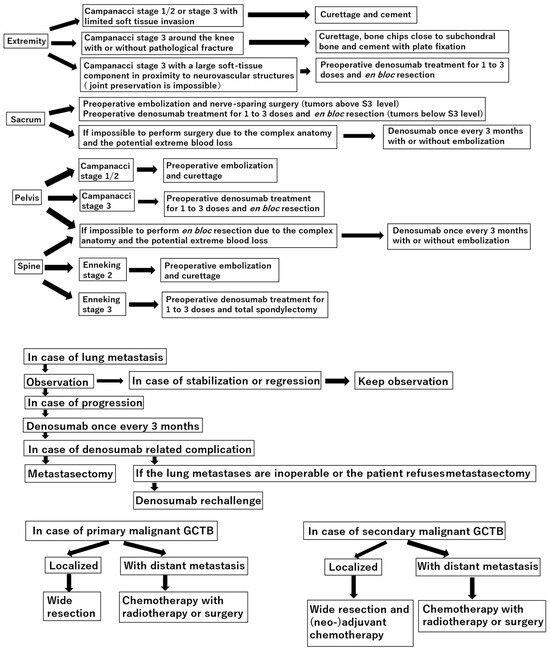
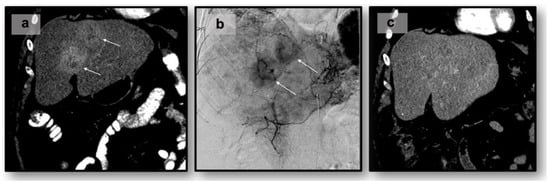
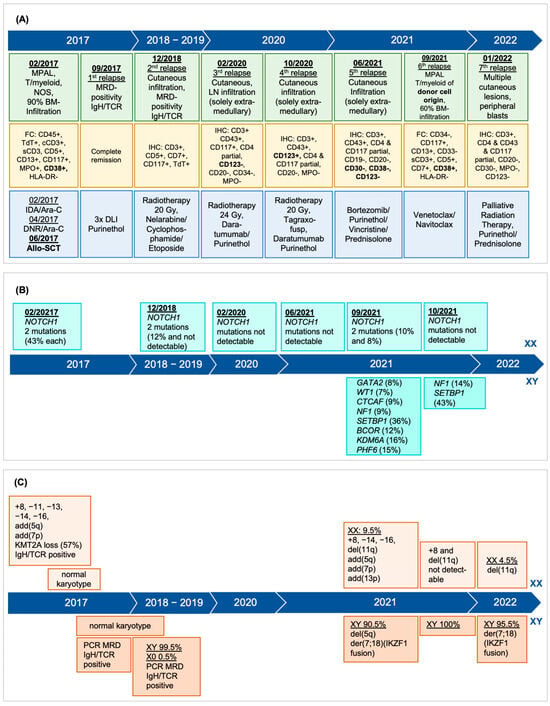
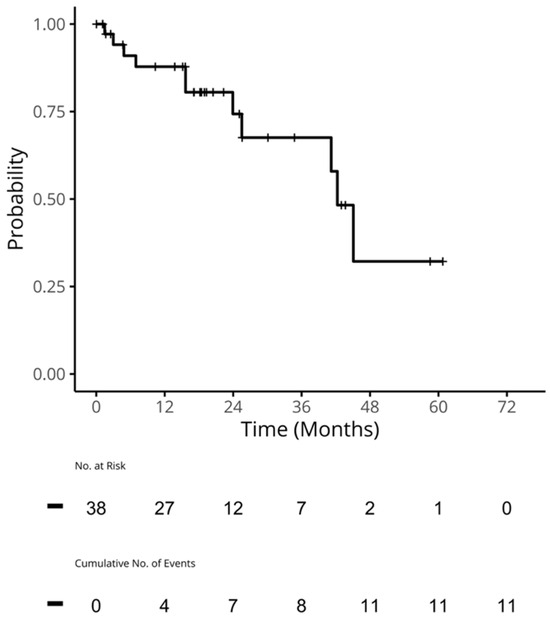
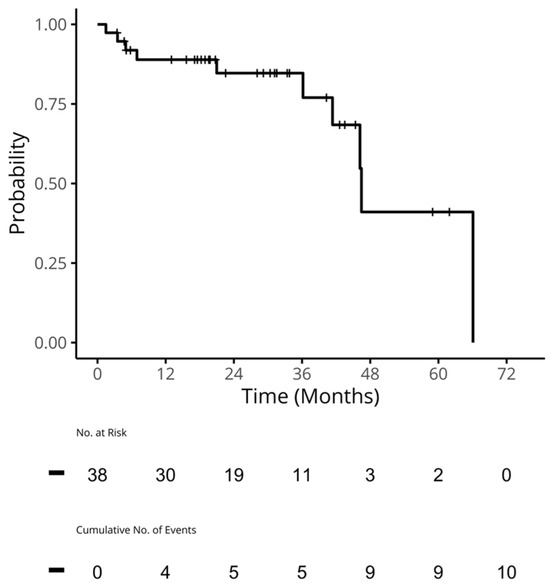
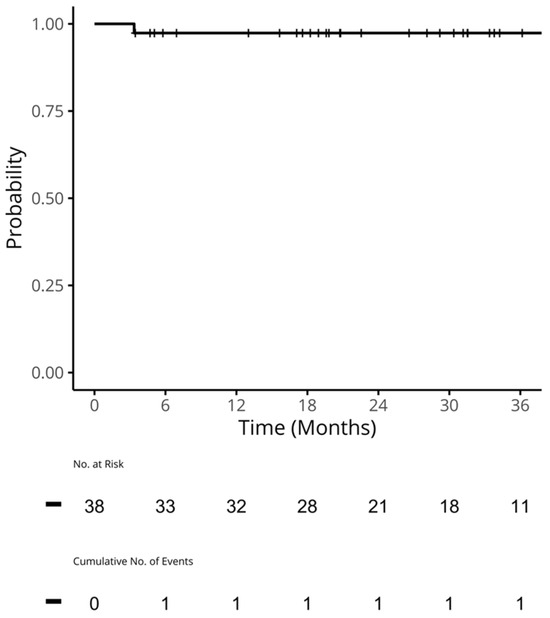
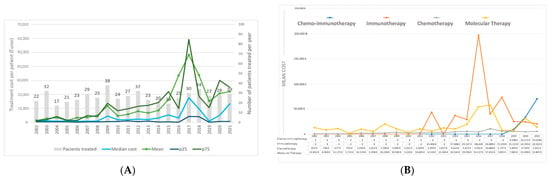
Figure 1









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}