


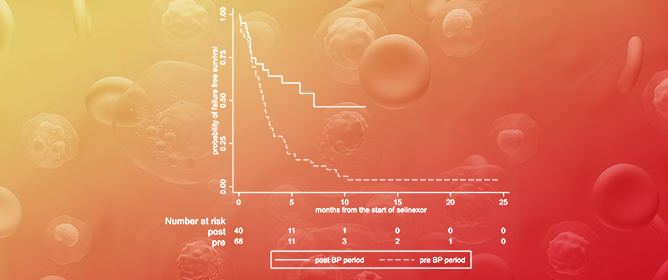
Curr. Oncol. 2024, 31(5), 2376-2392; https://doi.org/10.3390/curroncol31050177 - 23 Apr 2024
Abstract
Patient-reported outcomes (PROs) offer a diverse array of potential applications within medical research and clinical practice. In comparative research, they can serve as tools for delineating the trajectories of health-related quality of life (HRQoL) across various cancer types. We undertook a secondary data
[...] Read more.
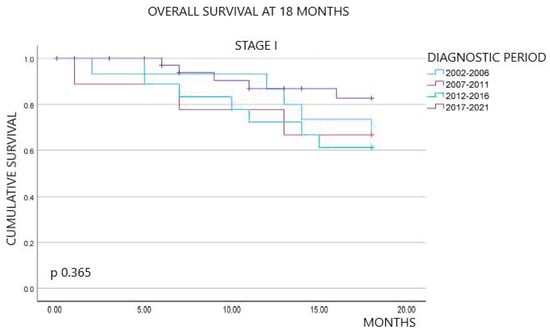
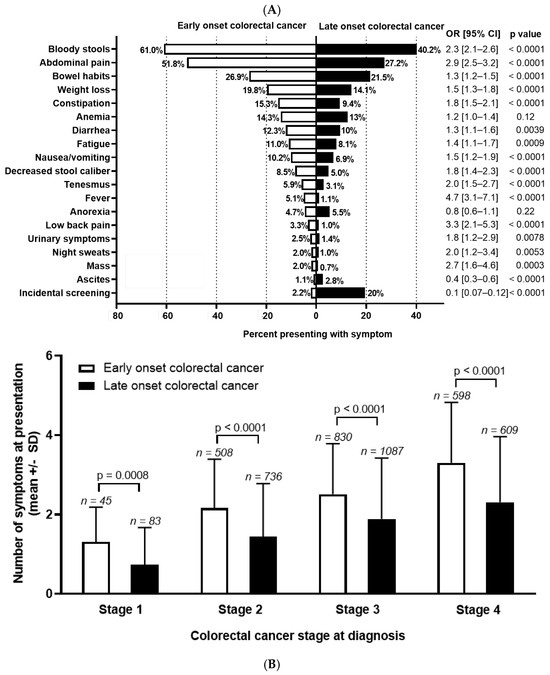
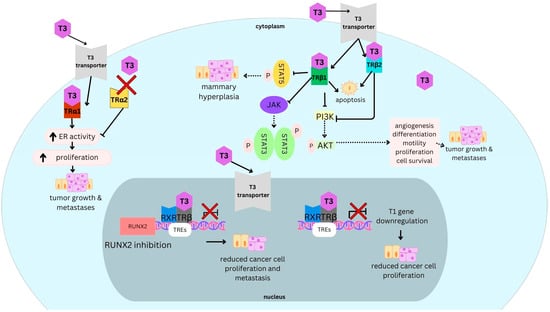
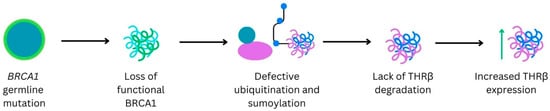
Patient-reported outcomes (PROs) offer a diverse array of potential applications within medical research and clinical practice. In comparative research, they can serve as tools for delineating the trajectories of health-related quality of life (HRQoL) across various cancer types. We undertook a secondary data analysis of a cohort of 1498 hospitalized cancer patients from 13 German cancer centers. We assessed the Physical and Mental Component Scores (PCS and MCS) of the 12-Item Short-Form Health Survey at baseline (t0), 6 (t1), and 12 months (t2), using multivariable generalized linear regression models. At baseline, the mean PCS and MCS values for all cancer patients were 37.1 and 44.3 points, respectively. We observed a significant improvement in PCS at t2 and in MCS at t1. The most substantial and significant improvements were noted among patients with gynecological cancers. We found a number of significant differences between cancer types at baseline, t1, and t2, with skin cancer patients performing best across all time points and lung cancer patients performing the worst. MCS trajectories showed less pronounced changes and differences between cancer types. Comparative analyses of HRQoL scores across different cancer types may serve as a valuable tool for enhancing health literacy, both among the general public and among cancer patients themselves.
Full article
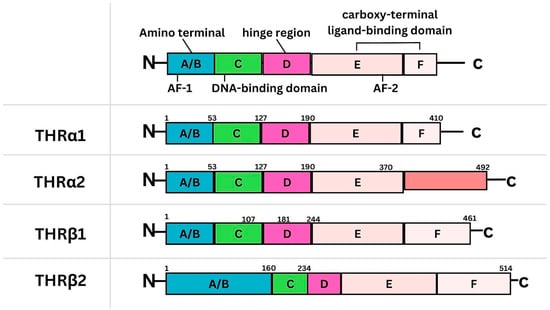
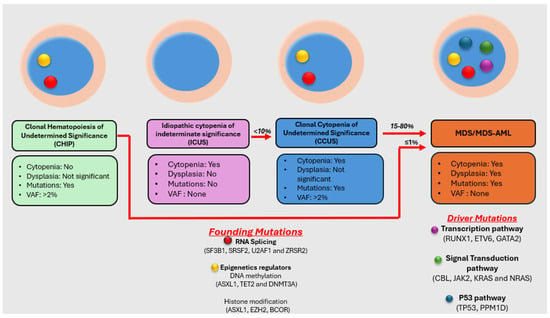





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}