


Curr. Oncol. 2024, 31(4), 2274-2277; https://doi.org/10.3390/curroncol31040168 - 17 Apr 2024
Abstract
Maintenance chemotherapy is a standard treatment in patients with non-progressive advance staged IV non-squamous non-small cell lung cancer after induction therapy. Here, we report the case of a 53-year-old man undergoing a maintenance monotherapy with pemetrexed who presented prolonged pancytopenia despite filgrastim injections.
[...] Read more.
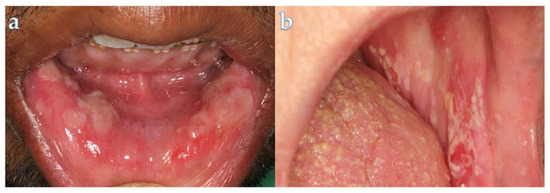
Maintenance chemotherapy is a standard treatment in patients with non-progressive advance staged IV non-squamous non-small cell lung cancer after induction therapy. Here, we report the case of a 53-year-old man undergoing a maintenance monotherapy with pemetrexed who presented prolonged pancytopenia despite filgrastim injections. A bone marrow aspiration revealed a macrophage activation syndrome with Leishmania amastigotes. A Polymerase Chest Reaction testing confirmed the diagnosis of visceral leishmaniasis. Treatment with liposomal amphotericin B was started. Oncologists should bear in mind that visceral leishmaniasis in endemic areas can potentially induce severe and prolonged pancytopenia in immunosuppressed patients, during chemotherapy in particular.
Full article
(This article belongs to the Section Thoracic Oncology)
►
Show Figures
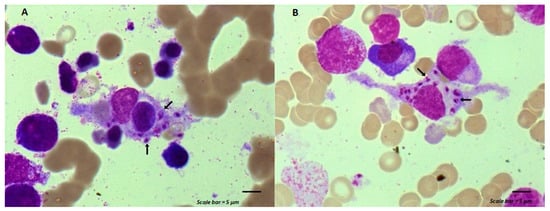
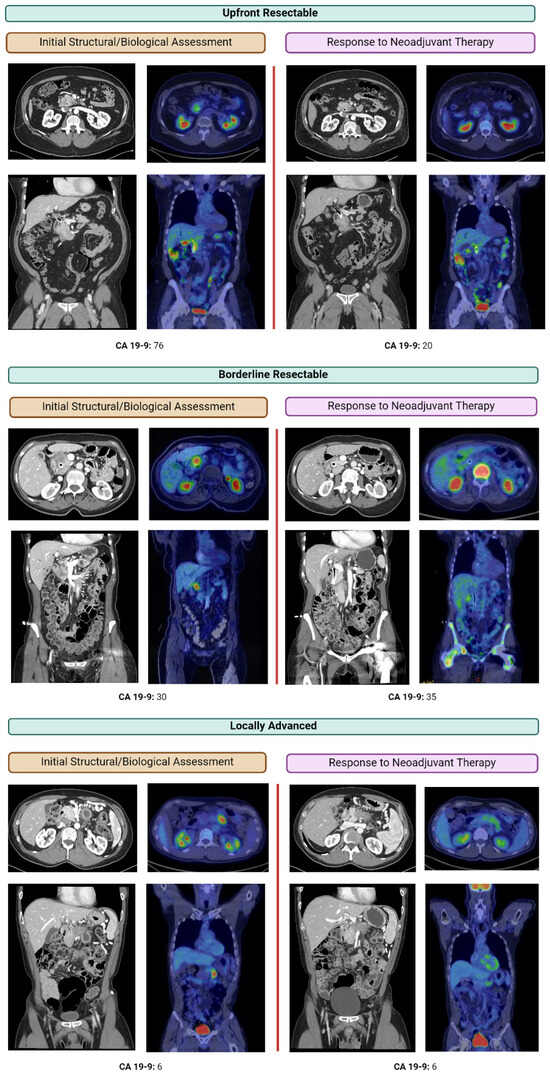
Figure 1









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}